

In this page you will find out more about YAMBO and its capabilities.
[00] Introduction
[01] Features
[01.01] Quasiparticle properties
[01.02] Spectroscopic properties
[01.03] Postprocessing
[02] Technical Details
[02.01] Methods and compatibility
[02.02] Platforms and parallel computing
[03] Reference publications
[03.01] Main YAMBO papers
[03.02] Papers describing YAMBO implementations

[00] Introduction
YAMBO is a plane-waves first-principles code for calculating excited-state properties – such as quasiparticle energies and optical spectra – of solid-state systems within the framework of many-body perturbation theory (MBPT) and time-dependent density functional theory (TDDFT).
Quasiparticle energies are calculated within the GW approximation for the self-energy. Optical properties are evaluated either by solving the Bethe–Salpeter equation (BSE) or by using the adiabatic local density approximation (ALDA).
YAMBO calculations require a previously computed electronic structure and for this reason it is currently interfaced with Quantum ESPRESSO and ABINIT.
Aiida plugins for YAMBO, such as YamboCalculation and YamboConvergence, automatise the very complex multi-parameter dependence that characterises GW calculations.
[01] Features
What can YAMBO do?
[01.01] Quasiparticle properties
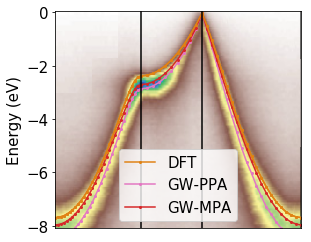
- GW calculations of electronic excitation
- Dynamical screening (plasmon-pole, multi-pole approximations or full-frequency)
- Finite temperature electronic energies and lifetimes
- Electron-phonon corrections
[01.02] Spectroscopic properties
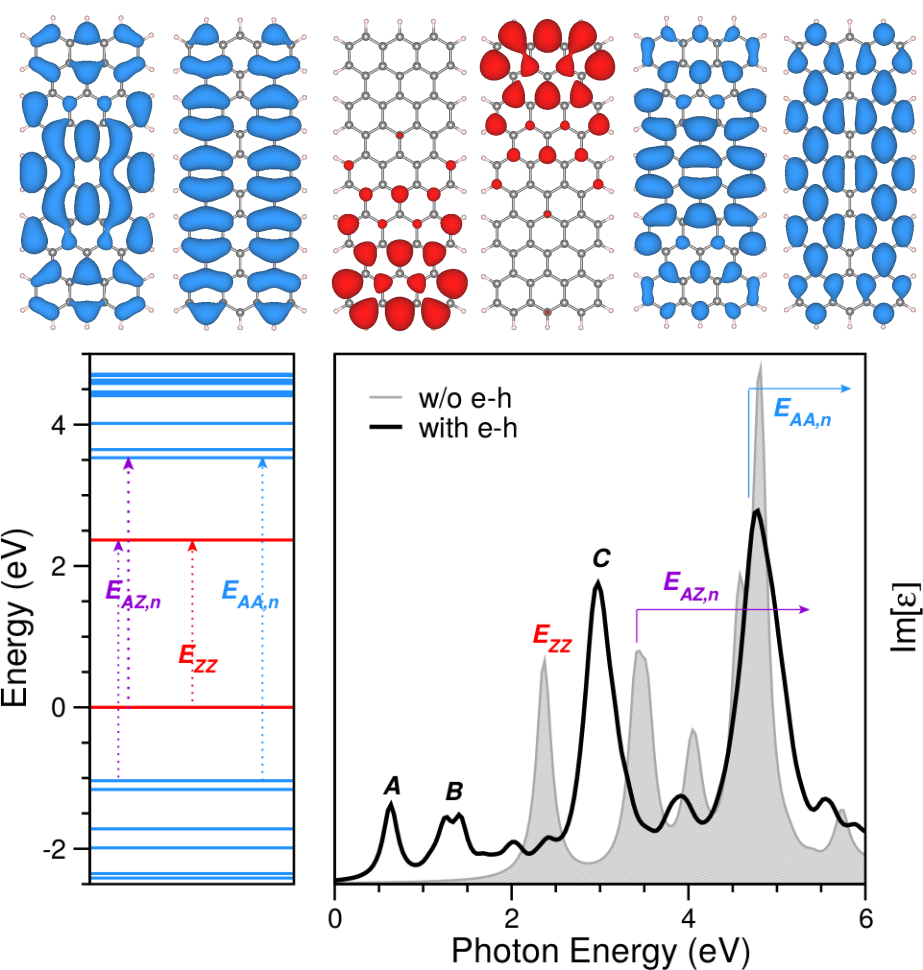
- Optical absorption and excitons
- Electron Energy Loss
- Kerr rotation
- Real-Time dynamics: Time–dependent Screened Exchange
- Nonlinear optical properties (including Berry’s phase polarization)
[01.03] Postprocessing
- Interpolation of quasiparticle and exciton energies and double grids
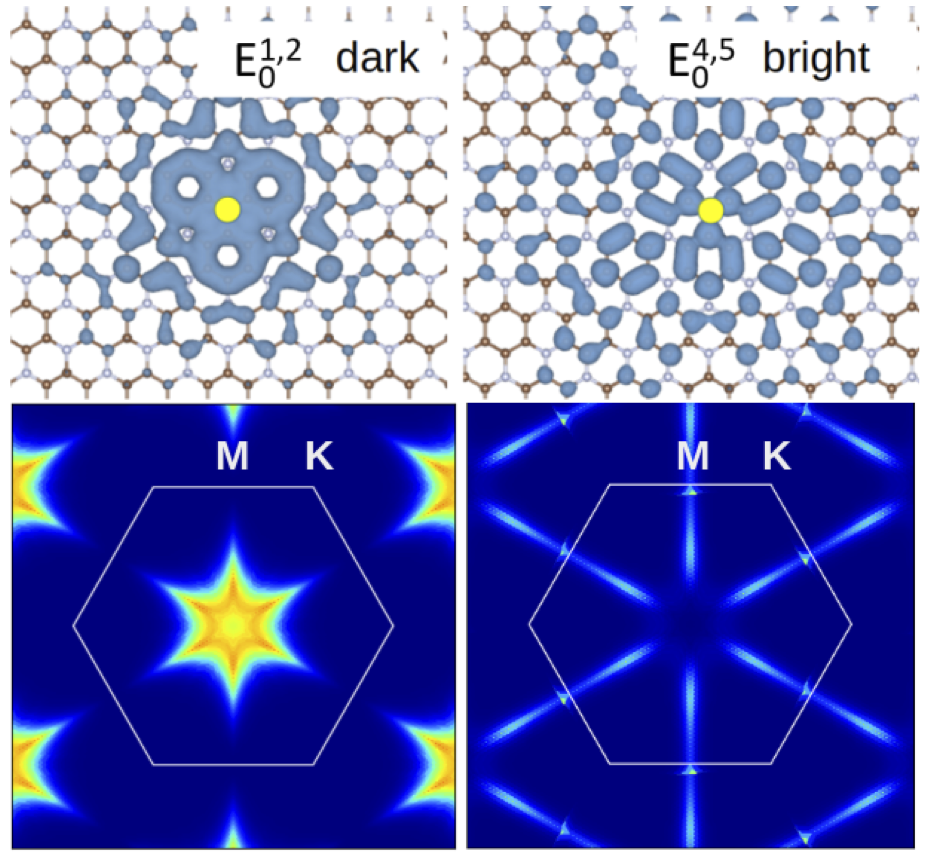
- Representation of excitonic wave functions in real and reciprocal space
- Python data analysis and scripting tool including:
- Quality-of-life automated scripts
- Visualization and plotting options for most quantities
- Data analysis tools for encoded databases beyond standard outputs
[02] Technical details
[02.01] Methods and compatibility
YAMBO supports norm-conserving pseudopotentials and many exchange-correlation functionals, from LDA to generalized-gradient corrections (PW91, PBE, B88-P86, BLYP) to meta-GGA, exact exchange (HF) and hybrid functionals (PBE0, B3LYP) via the libxc library.
Noncollinear magnetism and spin-orbit coupling calculations are also supported. There are several available Coulomb interaction cutoff geometries, sum-over-states terminators and interpolation schemes.
YAMBO is a Fortran/C code that exploits a number of optimized libraries such as BLAS, LAPACK, FFTW, SCALAPACK, NetCDF/HDF5, PETSC, and SLEPC.
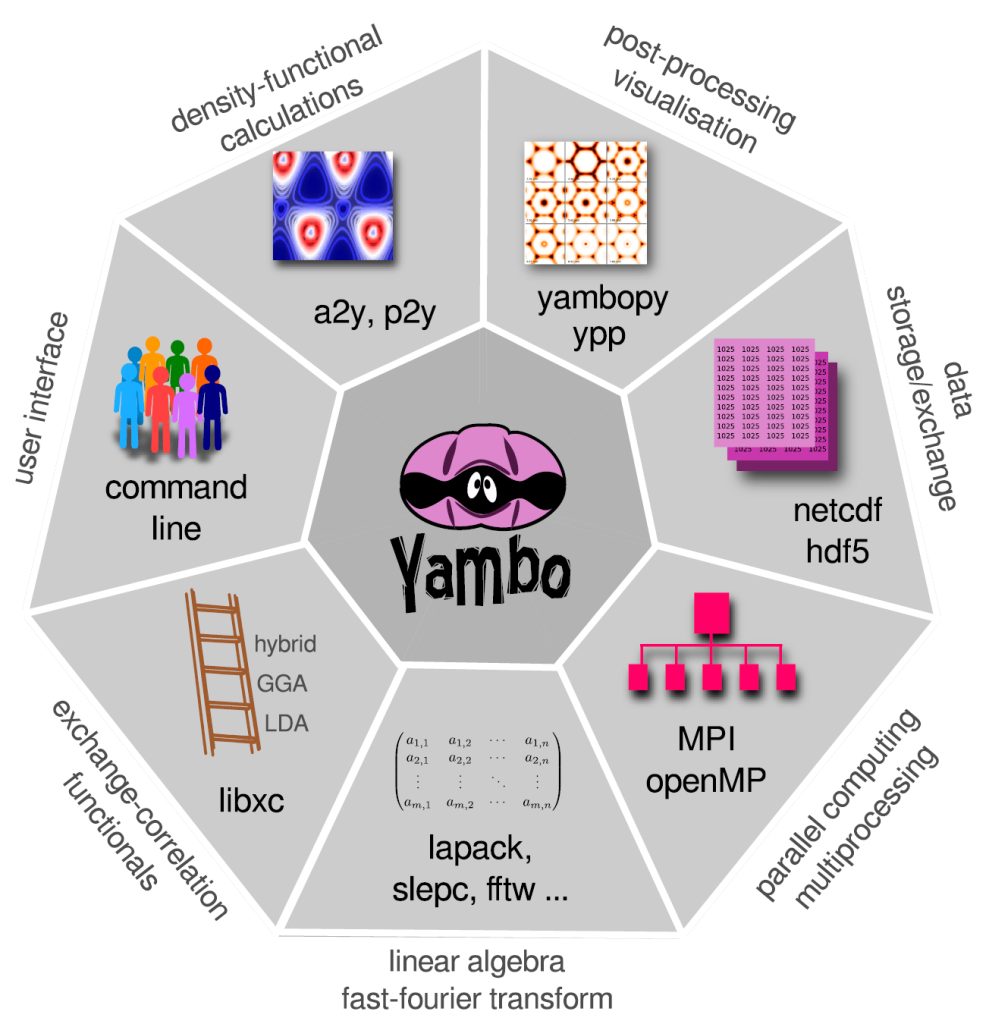
[02.02] Platforms and parallel computing
The code is parallelized over several MPI+OpenMP levels. YAMBO stores information in several database files (the biggest reaching few GBs in size for our systems) for which a NetCDF/HDF5 format is adopted, optimizing IO and data portability. The code has been extensively tested and used on different HPC architectures, for large scale systems.
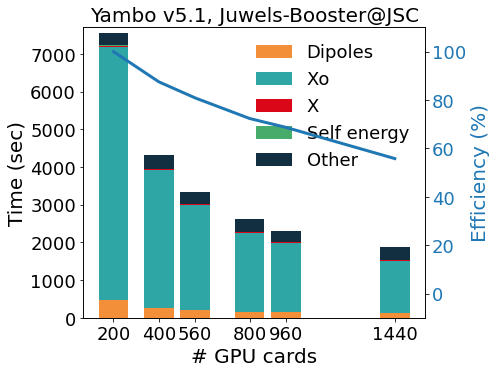
The YAMBO implementation of GW is parallel on the k/q grids, bands summation and quasiparticle energies, using a hybrid MPI-OpenMP approach. Explicit OpenMP support is implemented following different strategies according to the specific kernels. The BSE routines are parallel on k-points, electron-hole basis elements, and transitions. GW and BSE calculations are computationally expensive and, for complex materials and surfaces, can be performed only exploiting the resources offered by modern Tier0 systems.
The GPU porting was first made using CUDA-Fortran, and more recently enlarged to other programming models (like OpenACC and OpenMP5, both in development). We make an intense use of pre-processor macros that activate the language chosen at compile time, allowing YAMBO to optimally integrate MPI-OpenMP with programming models for GPGPU.
[03] Reference publications
[03.01] Main YAMBO papers
• Many-body perturbation theory calculations using the yambo code
D. Sangalli, A. Ferretti, H. Miranda, C. Attaccalite, I. Marri, E. Cannuccia, P.M. Melo, M. Marsili, F. Paleari, A. Marrazzo, G. Prandini, P. Bonfa’, M. O. Atambo, F. Affinito, M. Palummo, A. Molina Sanchez, C. Hogan, M. Grüning, D. Varsano and A. Marini
Journal of Physics: Condensed Matter 31 325902 (2019)

• yambo: An ab initio tool for excited state calculations
A. Marini, C. Hogan, M. Grüning, and D. Varsano
Computer Physics Communications 180, 1392 (2009)

[03.02] Papers describing the YAMBO implementations
- Efficient GW calculations via interpolation of the screened interaction in momentum and frequency space: The case of graphene
A. Guandalini, D. A. Leon, P. D’Amico, C. Cardoso, A. Ferretti, M. Rontani, and D. Varsano
Phys. Rev. B 109, 075120 (2024) - Efficient full frequency GW for metals using a multipole approach for the dielectric screening
D. A. Leon, A. Ferretti, D. Varsano, E. Molinari, C. Cardoso
Phys. Rev. B 107, 155130 (2023) - Towards high-throughput many-body perturbation theory: efficient algorithms and automated workflows
M. Bonacci, J. Qiao, N. Spallanzani, A. Marrazzo, G. Pizzi, E. Molinari, D. Varsano, A. Ferretti, D. Prezzi
npj Comput Mater 9, 74 (2023) - Efficient GW calculations in two dimensional materials through a stochastic integration of the screened potential
A. Guandalini, P. D’Amico, A. Ferretti, D. Varsano
npj Comput Mater 9, 44 (2023) - Frequency dependence in GW made simple using a multipole approximation
D. A. Leon, C. Cardoso, T. Chiarotti, D. Varsano, E. Molinari, and A. Ferretti
Phys. Rev. B 104, 115157 (2021) - Reproducibility in G0W0 Calculations for Solids
T. Rangel, M. Del Ben, D. Varsano, G. Antonius, F. Bruneval, F. H. da Jornada, M. J. van Setten, O. K. Orhan, D. D. O’Regan, A. Canning, A. Ferretti, A. Marini, GM Rignanese, J. Deslippe, S. G. Louie, J. B. Neaton
Computer Physics Communication 255, 107242 (2020) - Nonlinear optics from an ab initio approach by means of the dynamical Berry phase: Application to second- and third-harmonic generation in semiconductors
C. Attaccalite, M. Grüning
Phys. Rev. B 88, 235113 (2013) - Real-time approach to the optical properties of solids and nanostructures: Time-dependent Bethe-Salpeter equation
C. Attaccalite, M. Grüning, and A. Marini
Phys. Rev. B 84, 245110 (2011) - Exciton-Plasmon States in Nanoscale Materials: Breakdown of the Tamm−Dancoff Approximation
M. Grüning, A. Marini, X. Gonze
Nano Lett. 9, 2820-2824 (2009) - Exact Coulomb cutoff technique for supercell calculations
C. A. Rozzi, D. Varsano, A. Marini, E. K. U. Gross, and A. Rubio
Phys. Rev. B 73, 205119 (2006)