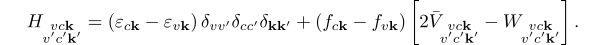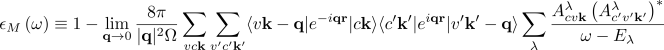I am planning to calculate the dielectric function of a given material with transferred momenta (i.e., q is not equal to 0) using the GW+BSE method. While reading the "BSE hBN Yambo Virtual 2021 version" (I really appreciate your hard work and selfless and detailed guidance), I noticed that all the BSE solvers seem to be performed under the condition of q approaching zero, rather than arbitrary transferred momenta.
I think the transferred momenta could be added by setting the corresponding parameter in the input files, but I don't know the specific operation. Should I set the 'BSEQptR' parameter in the BSE Hamiltonian or make corresponding settings in the input file of the BSE solver? By the way, can someone please tell me the format of BSEQptR, as I cannot find it in https://www.yambo-code.eu/wiki/index.ph ... s#QPkrange? For example, if I need to calculate N q-points, is the following format correct?
Code: Select all
% BSEQptR
1 | N | # [BSK] Transferred momenta range----------------------------------------
Liang
University of Science and Technology of China